
RENAL DISEASE - TOPUP
Coming up.... Case scenario Renal disease (video will be uploaded soon)
The Kidney
Each human kidney contains approximately 1 million functional units, called nephrons, which are primarily involved in formation. Formation ensures that the body eliminates the final products of metabolic activities and excess water in an attempt to maintain a constant internal environment (homeostasis).
Urine formation by each nephron involves 3 main processes:
-
Filtration at the glomerular level
-
Selective reabsorption from the filtrate passing along
the renal tubules
-
Secretion by the cells of the tubules into this filtrate.
Perturbation of any of these processes impairs the kidney’s
excretory function, resulting in azotemia, which is elevation of blood urea nitrogen (BUN) (reference range, 8–20 mg/dL) and serum creatinine (normal value, 0.7–1.4 mg/dl) levels.
The quantity of glomerular filtrate produced each minute by all nephrons in both kidneys is referred to as he glomerular filtration rate (GFR). Average GFR is about 125 mL/min (10% less for women) or 180 L/d. About 99% (178 L/d) is reabsorbed, and the rest (2 L/d) is excreted.
Measuring renal function
Radionuclide assessment of GFR is the criterion standard for measuring kidney function. However, because it is expensive and not widely available, serum creatinine concentration and creatinine clearance (CrCl) more commonly are used.
An inverse relationship between serum creatinine and GFR exists. However, the serum creatinine and CrCl are not sensitive measures of kidney damage for two reasons. First, substantial renal damage can take place before any decrease in GFR occurs. Second, a substantial decline in GFR may lead to only slight elevation in serum creatinine. An elevation in serum creatinine is apparent only when the GFR falls to about 60–70 mL/min. This is due to compensatory hypertrophy and hyperfiltration of the remaining healthy nephrons.
Because creatinine normally is filtered as well as secreted into the renal tubules, the CrCl may cause the GFR to be substantially overestimated, especially as kidney failure progresses because of maximal tubular excretion. More accurate determinations of GFR require the use of inulin clearance or a radiolabeled compound, such as iothalamate. In practice, precise knowledge of the GFR is not required, and disease process usually can be monitored by the estimated GFR (eGFR) using different methods, as shown below.
The CrCl is best calculated by obtaining a 24-hour collection for creatinine and volume and then using the following formula:
CrCl (ml/min) = U/P × V
where U is the 24 hour creatinine in mg/dL, P is the serum creatinine in mg/dL, and V is the 24 hour volume/1440 (number of min in 24 hour).
Using the 24 hour creatinine in grams and the serum creatinine in milligrams, CrCl (mL/min) = creatinine [g/d]/ serum creatinine [mg/dL]) × 70. An adequate 24 hour collection usually reflects a creatinine generation of 15–20 mg/kg in women and 20–25 mg/kg in men. When 24 hour creatinine is measured, the adequacy of the collection must be established prior to calculation of the creatinine clearance.
Estimation of GFR by formula derived from plasma creatinine

In this formula, the body weight that is used may substantially alter the derived GFR. In obese or edematous patients, the total body weight is much greater than the lean body mass from which creatinine is derived, and GFR is overestimated. In cachectic patients with depleted lean body mass, creatinine production is so low that serum creatinine is frequently less than 1.0 mg/dL and overestimates the true GFR. Robert and associates adjusted the Cockroft-Gault equation to incorporate ideal body weight from a nomogram and serum creatinine concentration corrected to 1.0 mg/ dl (if < 1.0 mg/dL). Using this modification they found that single measurements in hemodynamically stable patients correlate more closely with inulin clearance than either a 30 minute or a 24 hour creatinine clearance.
A more complex nomogram, based on a study called Modification of Diet in Renal Disease (MDRD), is independent of body weight:
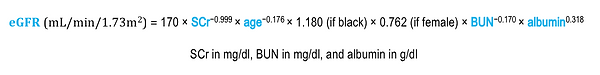
When renal function is rapidly changing, serum creatinine–based nomograms are subject to the same limitations as serum creatinine itself. In fact, serum creatinine levels do not begin to increase above normal until the GFR declines below 50 mL/min/1.73 m2, and occasionally they will remain normal even when the GFR dips as low as 20 to 40 mL/min/1.73 m2.
Intact nephron hypothesis
The intact nephron hypothesis describes the process of adaptation in chronic kidney disease. It states:
-
As CKD advances, kidney function is supported by a
diminishing pool of functioning (or hyper-functioning) nephrons, rather than a relatively constant number of nephrons each with diminished function.
-
The sum of the function of the damaged nephrons correlates with the total kidney GFR.
-
The chronically damaged kidney continues to regulate and responds to the needs of the organism in an organized, meaningful, and directed fashion.
Tubulo-glomerular feedback
Tubuloglomerular feedback may be a primary mechanism in renal autoregulation. When the GFR is increased, distal tubular NaCl delivery is enhanced. The increase in chloride is sensed by the macula densa, which triggers the release of renin from the adjacent afferent arteriole. Angiotensin is elaborated and arteriolar constriction ensues, which decreases the GFR. It may also be a compensatory mechanism to prevent polyuria in acute renal failure (“acute renal success”). When the thick ascending loop becomes ischemic, reabsorption of NaCl ceases, the ability of the tubule to concentrate urine is lost, and theoretically, intractable polyuria should result. Thurau and Boylan16 suggested that the increased delivery of NaCl to the macula densa triggers angiotensin-mediated arteriolar constriction, which decreases the GFR, induces oliguria, conserves intravascular volume, and protects the organism from dehydration—so-called acute renal success.
Renal autoregulation
Autoregulation enables the kidney to maintain solute and water regulation independently of wide fluctuations in arterial blood pressure. It is noteworthy that the urinary flow rate is not subject to autoregulation. Tubular water reabsorption determines the urinary flow rate and is closely related to hydrostatic pressure in the peritubular capillaries. Hypotension, whether induced or inadvertent, results in a decreased urinary flow rate that may be correctable only when arterial blood pressure is restored toward normal.
The precise mechanism of renal autoregulation is not yet defined. It is probably produced in part by a direct contractile response of the smooth muscle of the afferent arteriole to stretch. Nitric oxide (NO) may also be involved. At low perfusion pressures, angiotensin II also appears to play a role by constricting the efferent arterioles, thus maintaining the glomerular filtration rate. This is believed to be the explanation of the renal failure that sometimes develops in patients with poor renal perfusion who are treated with drugs which inhibit angiotensin-converting enzyme.
Renal vascular resistance appears to be mediated by variable resistance of the preglomerular afferent arteriole. As mean arterial pressure decreases, renal vascular resistance decreases and renal blood flow (RBF) is maintained. The most plausible explanation is a myogenic response in which the arterioles constrict in response to increased arterial pressure and vice versa. When arterial pressure increases through the autoregulatory range, it enhances delivery of sodium chloride to the cells of the macula densa, which induces afferent arteriolar constriction and decreased RBF and GFR. The opposite effect occurs when arterial pressure declines.
Though not abolished by most anesthetic agents, autoregulation appears to be impaired in severe sepsis, in acute renal failure, and possibly during cardiopulmonary bypass (CPB). In these situations, RBF declines strikingly during hypotension. It is restored by normalization of renal perfusion pressure, even if such normalization is achieved by vasoconstrictor therapy.
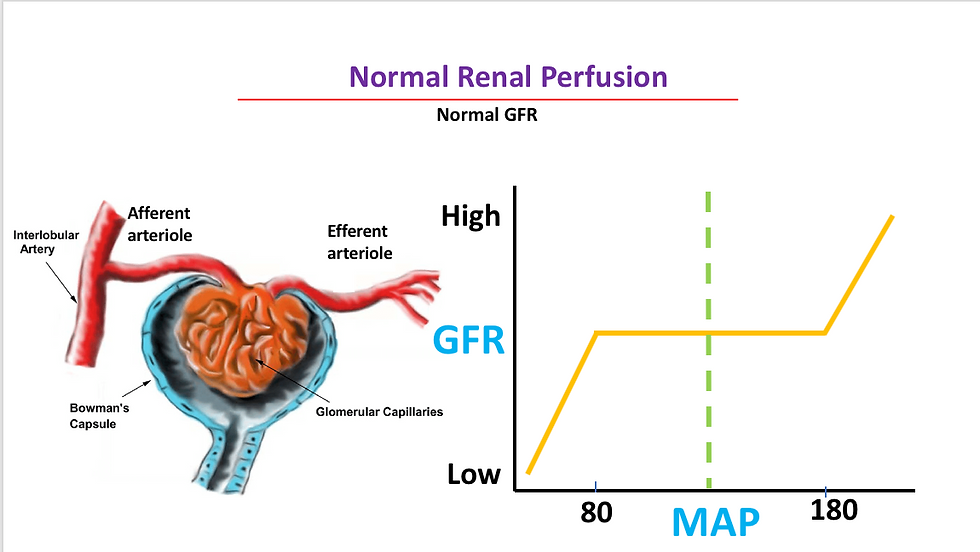
normal glomerular blood flow with normal glomerular filtration rate (GFR).
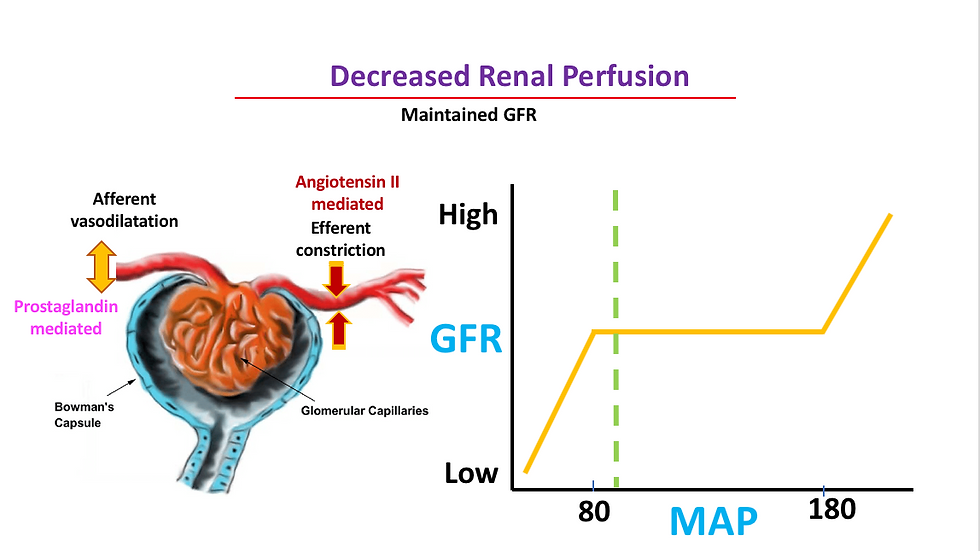
reduced renal perfusion pressure within the autoregulatory range, caused by intraoperative conditions such as anesthesia and medication induced hypotension or hypovolemia. normal GFr is maintained with prostaglandin-mediated afferent arteriolar vasodilation and angiotensin II–mediated efferent arteriolar vasoconstriction.
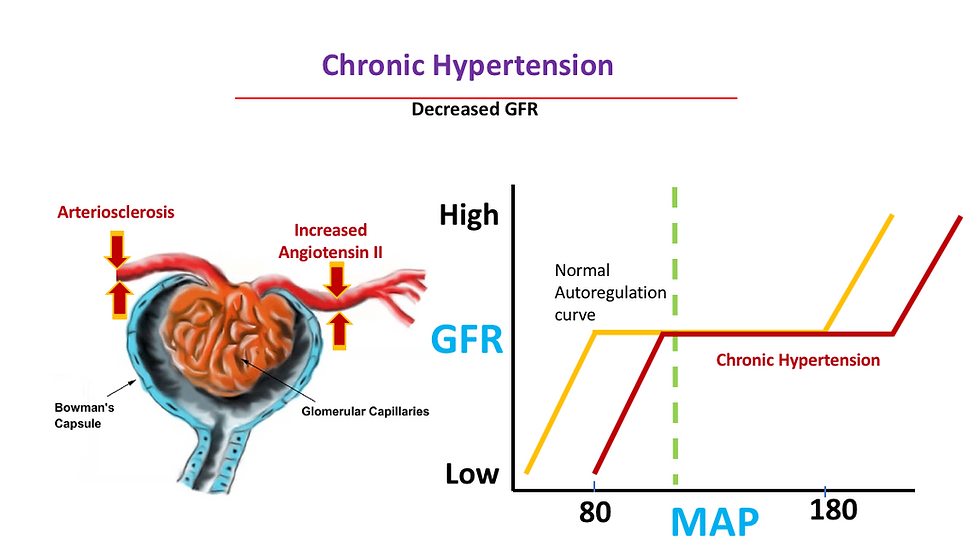
Effect of chronic hypertension on the preglomerular arterial vessels, primarily the afferent arterioles. Chronic hypertension eventually leads to thickening of arteriole walls and narrowing of lumen, a process known as arteriolosclerosis. This results in inadequate blood flow through the glomeruli and may produce glomerular and tubulointerstitial ischemia.
normal glomerular blood flow with normal glomerular filtration rate (GFR).
Renal Function Tests


Perioperative fluid management in a patient with anuric renal failure
Fluid management of the patient with anuric renal failure for nontransplantation surgery should avoid excessive intravascular fluid administration and ECF volume expansion and should maintain or correct electrolyte and acid-base status. The clinical anesthesiologist should also attempt to prevent precipitating conditions that would require dialysis during the immediate postoperative period (e.g. hyperkalemia, pulmonary edema, metabolic acidosis). Dialysis is difficult or impossible in the hemodynamically unstable patient, and anticoagulation may be strongly contraindicated for several hours postoperatively. Avoidance of hypotension may be indicated to maintain coronary or cerebral perfusion pressure. In patients with acute renal failure, hypotension may worsen ischemic renal injury. This contrasts with the lack of concern for an adverse effect on renal function in the patient with chronic renal failure. Recent dialysis often induces acute electrolyte shifts, hypovolemia, and acidosis. The best compromise appears to be dialysis 12 to 24 hours preoperatively. If dialysis is performed close to the time of surgery, volume removal should be minimal. Patients with renal failure are often malnourished, have hypoproteinemia, are anemic, and have poor glucose tolerance.
Hemodynamics should be closely monitored. If hypovolemia develops, colloid should be used early, but the physician should avoid producing vascular overexpansion. Likewise, interstitial fluid overload should be avoided because it would require acute postoperative dialysis. Crystalloid replacement of third-space losses should be limited to 1 to 2 ml/kg/hour, whereas blood loss should be replaced with colloid or RBCs. Correction of sodium, potassium, and acidosis can be achieved by the use of isotonic fluid without potassium, with reduced amounts of chloride, and increased amount of buffer, respectively. Initially, an infusion of 30% of calculated maintenance fluid rate is important because approximately 70% of normal fluid requirements are used in excreting solute through the kidney, a route no longer available. The pH and levels of sodium, potassium, bicarbonate, and glucose should be monitored at regular intervals.
CONTROVERSIES IN THE MANAGEMENT OF ACUTE RENAL FAILURE
“Renal Dose” of Dopamine
Despite more than thirty years of use as a renal vasodilator, low-dose dopamine (2μg/kg/min) has shown no evidence
of benefit in patients with acute oliguric renal failure. In fact, low-dose dopamine can have deleterious effects on hemodynamics (decreased splanchnic blood flow) immune function (inhibition of T-cell lymphocyte function) and endocrine function (inhibition of thyroid-stimulating hormone release from the pituitary).
Despite its widespread use, there is no evidence to support the use of low-dose dopamine to prevent or treat ARF, and, therefore, dopamine should be eliminated from routine clinical use for this indication. At first glance, it is difficult to understand why low-dose dopamine does not confer any benefit to patients with, or at risk for, ARF. Low-dose dopamine is effective in increasing renal blood flow and results in increased urine output. Clinicians frequently use “renal dose” dopamine in the hopes that such a maneuver might attenuate renal injury and improve survival. In addition, clinicians often interpret an increase in urinary output as proof that these two assumptions are valid. In reality, dopamine may increase urine output through five separate mechanisms.
Dopamine stimulates both dopaminergic and adrenergic (both α and β) receptors. As such, dopamine may affect renal blood flow by direct vasodilation (dopamine receptors), by increasing cardiac output (β-receptors) or by increasing perfusion pressure (α-receptors). At low doses, particularly, 2μg/kg/min, the dopaminergic effects tend to predominate, although wide variability seems to exist across patients and clinical conditions. In appropriate clinical settings, any of the mechanisms might increase effective renal plasma flow and thus increase urine output. Under such conditions, the increase in urine output might well be indicative of improved renal function. However, dopamine may also increase urine output by both inhibition of aldosterone release as well as inhibition of sodium-potassium ATPase at the tubular epithelial cell level. The effect of these last two mechanisms is to increase sodium excretion and thereby diuresis. Thus, apart from the direct and indirect effects on the renal vasculature, dopamine increases urine output because it is a diuretic.
Much of the enthusiasm for the use of this agent comes from the belief that dopamine increases renal blood flow and that such an outcome is desirable. Given that ARF in the critically ill is usually caused by acute tubular necrosis (ATN), it seems paradoxic that an agent that improves renal blood flow is not beneficial. Clearly, the continued popularity of so-called “renal dose” dopamine is the result, in large part, of this seemingly strong physiologic rationale even in the face of multiple negative clinical trials.
First, most clinicians use so-called “renal-dose” dopamine in the belief that they can improve renal blood flow and that this will restore oxygen delivery to portions of the kidney that are dysoxic. However, it is not clear that ischemic ATN is always the result of a decrease in renal blood flow nor is it clear that increasing renal blood flow will reverse or prevent this injury. The renal medulla is constantly on the brink of dysoxia owing to high demand and low delivery of oxygen. Although dopamine increases outer medullary blood flow in hypovolemic animals, it fails to improve outer medullary dysoxia. In fact, analogous to the situation with radiocontrast, the increased solute delivery to the distal tubular cells produced by the natriuretic effects of dopamine (via inhibition of proximal tubular resorption) might actually increase medullary oxygen consumption and thereby increase the risk of ischemia rather than decrease it. This aspect of renal physiology could explain why agents that increase renal blood flow are not protective. Indeed, this scenario would predict that such agents would actually be deleterious. In fact, two recent studies suggest that dopamine worsens renal tubular injury secondary to radiocontrast agents and in postcardiac surgery patients despite an increase in renal blood flow.
Finally, drugs that oppose dopamine activity, such as metoclopramide or haloperidol, are not associated with any adverse renal effects despite widespread use in the same populations where “renal dose” dopamine is often advocated. Indeed, both of these agents effectively negate any the renal vascular effects of low-dose dopamine, yet neither is associated with ARF. Thus, the existing experimental evidence does not provide conclusive support for dopamine and some studies raise concerns that dopamine may even be harmful. These studies also call into question the appropriateness of increasing urine output in the first place.
In the case of other types of diuretics, converting oliguric to nonoliguric renal failure may provide certain management advantages (e.g. controlling volume overload
and hyperkalemia) but does not affect overall renal recovery or outcome. Accordingly, on the basis of existing evidence, the use of dopamine to improve renal function cannot be recommended.
Overall, the physiologic considerations and the considerable clinical data amassed to date argue strenuously against the routine use of “renal dose” dopamine.
Role of frusemide in acute renal failure
Frusemide is a loop-diuretic, which inhibits Na+/2Cl-/K+ cotransport system in the luminal membrane of ascending limb of loop of Henle and thus promotes diuresis, and also promotes renal vasodilatation. Clearly ineffective in established oliguric ARF,28 it was able to exert protective effects when given before a potential renal insult in experiments involving isolated perfused rat kidneys. In these experiments, it was shown that modification of transport activity in the thick ascending limb of Henle by frusemide reduced the severity of renal damage. By reducing active NaCl transport, frusemide reduces oxygen requirement and may thus confer renal protection. By augmenting tubular blood flow, frusemide decreases the concentration of nephrotoxins and prevents tubular obstruction.
The clinical trials of frusemide as a renal rescue agent in established oliguric ARF have been generally disappointing. While frusemide established a polyuric state, it did not improve renal function, the requirement for dialysis or outcome. Continuous infusion of frusemide was detrimental to postoperative renal function.
The authors, Lassnigg and associates felt that this was a consequence of the activation of the sympathetic and the renin—angiotensin systems by frusemide, which in turn led to an increase in peripheral vascular resistance, and thus a decrease in cardiac index and renal perfusion. In another study, higher doses of frusemide were associated with renal dysfunction.
Ho and Sheridan examined the effectiveness of frusemide in their meta-analysis of nine studies, three of which were used to prevent ARF, and six to treat ARF. Most of the studies used frusemide at 1–2.5 mg/hour to prevent ARF, and 600–3400 mg/day in established ARF. There was no improvement in in-hospital mortality, requirement for hemodialysis, or proportion of patients with persistent oliguria. High-dose frusemide used in established ARF was associated with an increased risk of temporary deafness and tinnitus, and an increase in hospital stay. While it has been argued that frusemide may convert oliguric ARF into a
polyuric state, they felt that responders to frusemide might represent those with less-severe ARF. They concluded that frusemide was ineffective in the prevention or treatment of ARF in adults .
With regard to cardiac surgery, the study published in European Journal of Cardio-Thoracic Surgery cautions regarding a trend to potentially deleterious short-term nephrotoxic effects of postoperative frusemide infusion following high-risk cardiac surgery.